在美国,对于没有合法上市对比产品的器械,即使属于中低风险,仍然无法通过510(k) 申请途径建立实质等同,获得上市许可。针对这类产品,FDA于多年前建立了De Novo申报途径,对产品进行普通控制或特殊控制,避免按照最高类别III类申报,给企业降低负担,以便患者能够及时获得安全有效的器械。
De nove 途径于1997年加入到食品和药物管理局现代化法案(Food and Drug Administration Modernization Act, FDAMA)中,为在进行上市前通告510(K) 申请时收到“非实质等同”(NSE)而自动归为III类的低到中等风险的新型器械提供一个新的流程,任何企业在进行510(k)申请的时候收到NSE通知后30天内,都可以向FDA提交De Novo请求,根据风险评估对器械分...
FDA De Novo(产品分类)办理条件
在美国,对于没有合法上市对比产品的器械,即使属于中低风险,仍然无法通过510(k)
申请途径建立实质等同,获得上市许可。针对这类产品,FDA于多年前建立了De Novo申报途径,对产品进行普通控制或特殊控制,避免按照最高类别III类申报,给企业降低负担,以便患者能够及时获得安全有效的器械。
De nove 途径于1997年加入到食品和药物管理局现代化法案(Food and Drug Administration Modernization Act, FDAMA)中,为在进行上市前通告510(K) 申请时收到“非实质等同”(NSE)而自动归为III类的低到中等风险的新型器械提供一个新的流程,任何企业在进行510(k)申请的时候收到NSE通知后30天内,都可以向FDA提交De Novo请求,根据风险评估对器械分类成I类或者II类。
2012年7月9日,根据食品和药品监督管理局安全和创新法案(Food and Drug Administration Safety and Innovation Act, FDASIA)607章修订的FD&C法案的513 (f) (2)章节,为企业提供了一个新选择,如果企业已经确定没有与他们的产品已上市的等价器械,不需要先提交510 (k) 申请,可以直接向FDA提交De Novo请求,根据风险评估对器械分类成I类或者II类。
该申请途径实施至今,已经有235种器械通过此途径上市,其中2012年以后有170种。
FDA本次发布的De Novo分类规则新草案更加透明有效地阐明了在该路径中提交文件的需求和FDA审核的过程,例如,拟议的法规和要求将提供关于重新分类过程的结构、清晰度和透明度,包括与重新分类请求的格式和内容有关的要求,以及接受、批准、拒绝和撤回重新分类请求的过程和标准。如果最终确定,将有助于对新型医疗设备进行适当分类,使设备开发商能够利用这一路径开发更多的新型设备。
De Novo路径使得更多符合现代性能安全标准的新型器械上市,并可以作为510 (k) 申请路径实质等同评价的对比产品,有利于FDA正在进行中的510 (k) 改革。与此同时,FDA也会采取新的方法,促进在510(k)路径的实质等同对比过程中使用更加现代的对比产品,从而又促进更多的器械采用De Novo途径。
FDA已经发布新的De Novo指南,指南将帮助FDA更好的监管新技术,既能够保护患者,同时促进创新技术改善人们的健康状况。
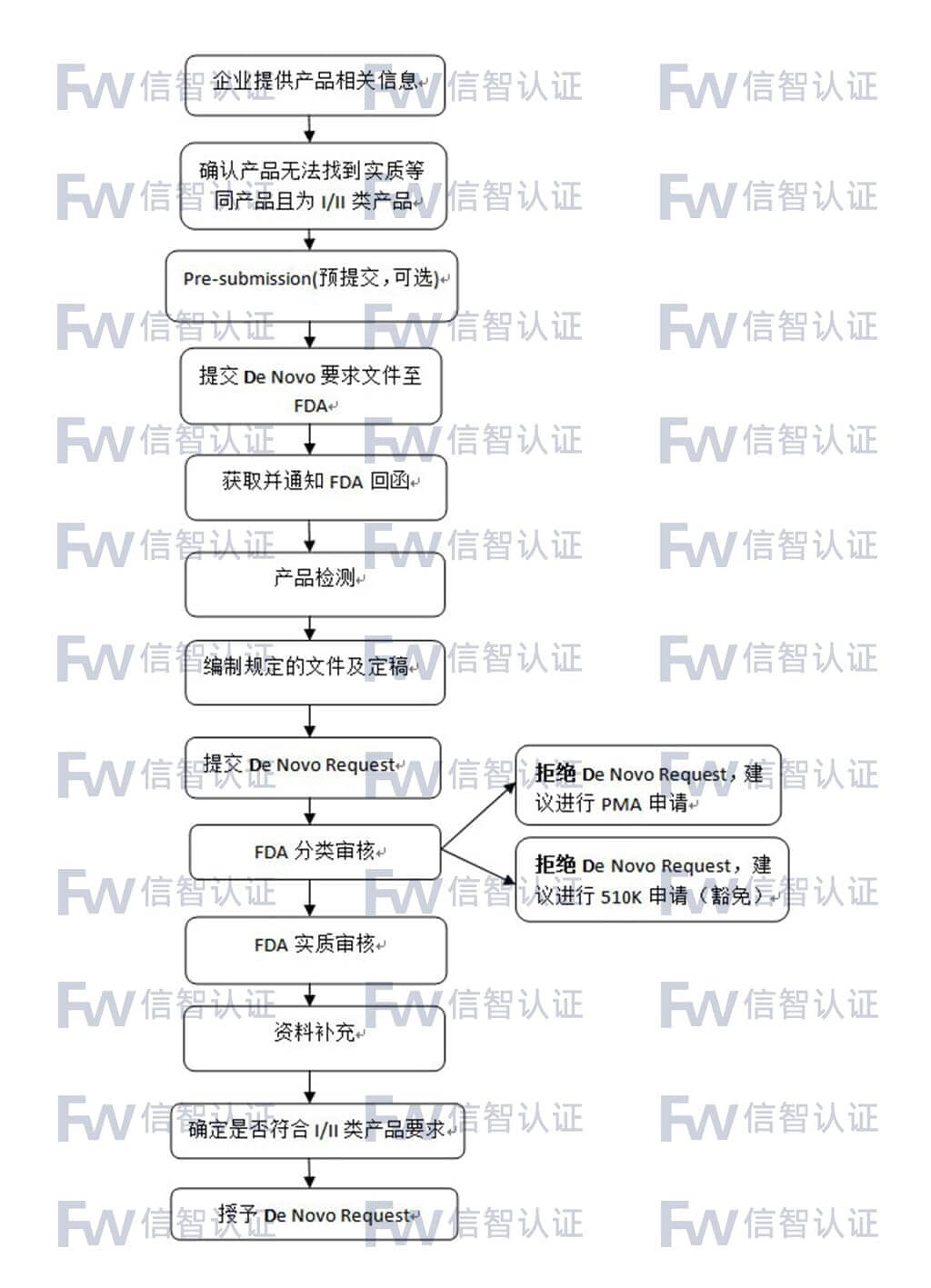
De Novo Request申请适合于当前无法找到实质等同产品且其安全和有效性可通过一般和特殊控制来保证的产品。鉴于这类申请的特殊性,FDA建议在正式提交前先进行预提交(Pre-submission)。
信智认证相关服务
确认产品是否符合De Novo Request
准备De Novo Request申请资料
资料递交与进度跟踪
FDA De Novo(产品分类)行政时间
| 名称 | 时限 |
|---|---|
| Pre-submission(可选)资料审核 | FDA官方时间(天):90天 |
| De Novo classification资料审核 | FDA官方时间(天):120天 |
FDA De Novo(产品分类)咨询服务流程